
Introduction
Reverse phase chromatography is a technique widely used to purify peptides. This technique owes its popularity to the fact that the purification times are quite short and the efficiencies achieved. The mobile phases used (mainly acetonitrile and water) are volatile which facilitates the concentration / lyophilization steps.
Sample preparation – peptides’s solubility
The solubility of the peptides in solution depends on several parameters:
- The amino acid sequence (if hydrophobic residues are present, this reduces the solubility in an aqueous medium)
- The size and the tertiary structure of the peptides. The solubilization can change depending on the distribution of the residues
- Percentage of residues loaded (it will be necessary to apply particular pH to help solubilization).
There are some rules to guide in the solubilization in aqueous medium:
- Peptides consisting of less than 5 residues are generally soluble unless the residues are hydrophobic (tryptophan, isoleucine, leucine, phenylalanine, methionine, valine or alanine).
- Hydrophilic peptides containing more than 25% of charged residues (glutamic acid, asparagic acid, lysine, arginine and histidine) and less than 25% of hydrophobic residues are generally dissolved in an aqueous medium, provided that the charged residues are distributed in the whole sequence
- Hydrophobic peptides containing from 50% to 75% of hydrophobic residues may be insoluble or partially soluble in aqueous solutions, even if they contain 25% of charged residues (generally, they are soluble in TFA and formic acid
- Acid peptides (residues Glutamic acid and Asparagic acid) and basic (residues Ariginine, Lysine, Histidine) are more soluble at neutral pH than acidic pH.
- The highly hydrophobic peptides that have more than 75% hydrophobic residues do not dissolve in aqueous solutions, it is preferable to make a solid deposit to properly inject these peptides.
Peptide sequences comprising a very large proportion of Serine, Threonine, Glutamic acid, Asparagic acid, Lysine, Arginine, Histidine, Asparagine, Glutamine or Tyrosine may form an intermolecular hydrogen bond network and have a tendency to form gels in aqueous solution. When they are concentrated, these peptides can be treated as hydrophobic peptides.
Of course, before passing the sample on the column, it is preferable to test the solubility of a small part under the method start conditions. It’s better to choose a solvent easily removable by lyophilization to recover the solutes as pure as possible.
How are the peptides eluted?
The interactions that manage reverse-phase peptide purifications are between the stationary phase and the hydrophobic scaffold of the peptide. They are non-dispersive and low-selective with 2 main mechanisms that are adsorption and sharing.
Small peptides (with molecular weight less than 3kDa) are eluted by a sharing mechanism while the larger peptides are eluted mainly by an adsorption mechanism. That is to say that the molecules at the injection “sticks” against the stationary phase. Then they are eluted when the proportion of organic solvent reaches a defined percentage.
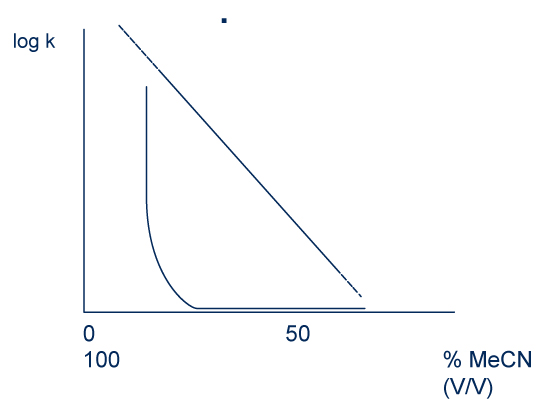 Retention according to the volume fraction of acetonitrile
Retention according to the volume fraction of acetonitrile
Choice of the stationary phase
 The choice of the stationary phase depends on the length of the peptide chains of the solutes which can limit the interactions by steric genes.
The choice of the stationary phase depends on the length of the peptide chains of the solutes which can limit the interactions by steric genes.
A C18 column will preferably be used for peptides of small and medium size, followed by a larger C8 peptide and C4 for polypeptides.
The second parameter to take into account is the porosity of the phase to be used. In fact the longer the peptide will be, the harder it will be to pass it into a small pore. Peptides from 1 to 5kDa will separate with a pore size of 100A, those of size between 5 and 20 kDa can be separated with a pore size of 200A and the polypeptides will be used with a pore size of 300A.
The last point affects the size of the particles. Given the difficulty of some purifications, it is not interesting to use sizes greater than 30µm.
Choice of solvent conditions
The most used solvents are water and acetonitrile. Acetonitrile allows to increase the elution of peptides. The fact that this solvent does not respond in UV and is easily removable, makes it an obvious choice.
It is common to use at the same time trifluoroacetic acid (TFA) as an ion-pairing agent which makes it possible to reduce the ionic interactions between the peptides and the stationary phase. Moreover, the high volatility of the TFA makes it a good element as easily removable (be careful, TFA must be present in the same proportion in the two solvents). The disadvantage of this product is that it will make the detection of MS difficult. It is then necessary to lower its concentration but it degrades the profile. As an alternative, you can use another agent such as formic acid that allows detection.
For applications with very hydrophobic compounds it is possible to use isopropanol. This solvent with a greater eluent power has another advantage, not negligible, keeping the biological activated of the peptides. This solvent is mainly used for polypeptides on C4 300Å column.
Generally we start the method with 5% of strong solvent, to increase the eluent force of 1% / min, which allows to have a better resolution.
For applications with polar peptides, use a HILIC mode.
Influence of some parameters
Several parameters are important to optimize the purifications:
- Gradient: As explained in the mechanism part, the peptides (> 3kDa) are eluted by an adsorption mechanism. A step gradient for elution may be useful.
- ion matching reagent. TFA is the most used but it is possible to use FA, PFPA, HFBA (anionic agent) as well as TEA, TBA (cationic agent) which can then help the separation of peptides with basic residues.
- Temperature: the temperature of the column is an optimization parameter. It reduces the viscosity of the solvent (allowing a significant reduction in back pressure) while allowing to reduce the time of release of the products. Be careful, however, not to degrade the solutes with too high temperature.
- Flow: If for the small molecules the flow is an optimization parameter allowing to save time with a small loss of resolution, for the more important peptides it is quite the opposite : an increase of the flow will cause a degradation of the profile of purification.
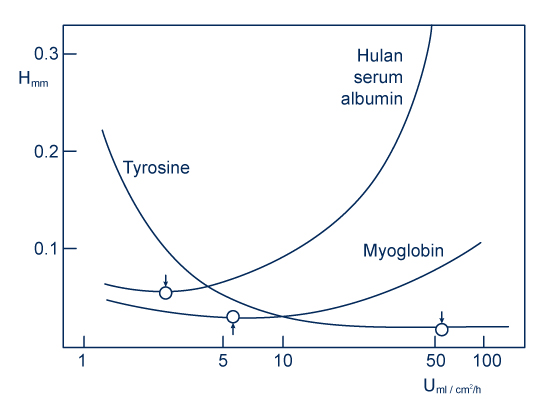 For peptides of low MW, there is an optimal flow (Van Deemter curve ….)
For peptides of low MW, there is an optimal flow (Van Deemter curve ….)
For peptides of high MW, the rise in flow alters the profile of the peaks. It can even cause peak duplication if the flow rate is too high.
Tyrosine: 181 g/mol
Myoglobin: 17 000g/mol
Albumin: >60 000g/mol
| If no retention | Retention too high or no elution |
Improve selectivity |
| Check the solvent of the sample (the eluting force must be less than or equal to the eluting force of the mobile start phase of run)
Check the concentration of the ion-pair reagent (usually the reagent is volatile) Decrease the concentration of organic solvent Decrease gradient slope |
Check the nature and concentration of the matching reagent
Increase organic solvent concentration Increase temperature Change column (test column C8 or C4 if C18 does not work) |
Modify the ion pairing reagent
Modify gradient slope Modify the organic solvent (for example mixture Acetonitrile / IPA, 90/10 max, replacing Acetonitrile alone for very hydrophobic peptide) Changing column (C18, phenyl, C8, C4) |
Find out more:
- Check out our column selection guide
- Discover our column list
- Discover our purification systems dedicated to peptides.














{kind=link}